{kind=link}

infantile polycystic kidney disease
coronal views of the fetal abdomen reveal large echogenic kidneys. a small bladder is noted along with less than average amniotic fluid implying diminished but not absent renal function at this point in pregnancy. the hypoechoic renal pelvis is clearly seen contrasted by the hyperechoic renal parenchyma.
infantile polycystic kidney disease
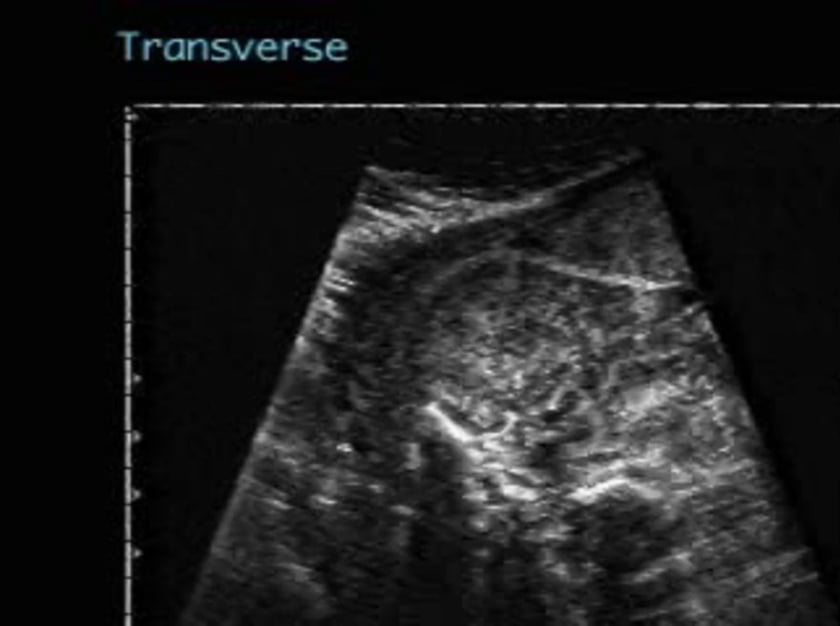
sagittal views demonstrate bilateral massively enlarged echogenic kidneys. the renal cortex is hypoechoic relative to the internal hyperechoic medullary area. the transverse view shows the abnormal size of the kidneys as they occupy greater than one third of the abdominal circumference. there is no evidence of bladder filling and oligohydramnios is evident.